- COMPLEXES (chimie)
- COMPLEXES (chimie)On appelle «complexe» tout édifice chimique formé par l’association de deux ou plusieurs entités chimiques indépendantes, ions ou molécules. C’est l’application à la chimie du concept selon lequel on désigne par «complexe» tout ce qui réunit plusieurs éléments différents.La chimie supramoléculaire en fournit de nombreux exemples en étudiant les phénomènes d’association et de reconnaissance intermoléculaires ou en synthétisant des molécules «creuses» capables de complexer sélectivement des anions ou des cations.Les exemples de complexes en chimie sont très nombreux et variés: c’est ainsi que l’ion PbC1+ est un complexe formé par le cation Pb2+ et l’anion C1-; le tri-iodure I-3 est constitué d’une molécule d’iode et d’un ion I-. FeS4+ est un complexe du fer ferrique Fe3+ et de l’anion sulfate S42-. On remarquera que S42- est lui-même un complexe de trioxyde de soufre S3 et de l’ion oxyde 2-.Toutefois, les complexes les plus importants dans la pratique sont ceux qui associent un cation métallique et une ou plusieurs entités «complexantes» appelées coordinats ou ligands. Les coordinats peuvent être des molécules organiques ou inorganiques ou des anions, ou assez rarement des cations comme l’ion nitrosyle +. C’est à ces complexes métalliques que nous limiterons notre propos.1. DéfinitionsDès 1798, Tassaert observa que les solutions ammoniacales de chlorure de cobalt bivalent laissent déposer, au bout de quelques heures, des cristaux orangés CoCl3, 6NH3 renfermant six molécules d’ammoniac pour une molécule de chlorure de cobalt trivalent; l’ammoniac est si fortement lié qu’il est possible de chauffer ce composé à 180 0C sans en perdre. Ce complexe se formule en fait [Co(NH3)6]Cl3, chlorure d’hexa-ammine cobalt (III). Mais il fallut attendre un siècle pour comprendre les curieuses propriétés de telles combinaisons minérales d’ordre supérieur, résultant de l’association de coordinats en nombre supérieur à celui fixé par la valence normale de l’ion métallique. En effet, la théorie de la coordination fut proposée en 1893 par Alfred Werner (prix Nobel de chimie en 1913) dont les travaux permirent de développer rapidement l’aspect structural conduisant à l’étude de nombreux cas d’isomérie et de complexes minéraux doués d’activité optique. Ces résultats ayant été obtenus bien avant l’énoncé de la théorie électronique des atomes, d’autres concepts vinrent ensuite expliquer la liaison de coordination. Ces approches successives (théorie des liaisons de valence dirigée, théorie du champ cristallin, théorie des orbitales moléculaires) aboutissent aujourd’hui à la notion du champ des coordinats, capable d’interpréter de manière satisfaisante la plupart des propriétés magnétiques, spectroscopiques, structurales et thermodynamiques des complexes métalliques (cf. COORDINATION [chimie] - Composés de coordination).Dans la plupart des cas, les coordinats sont des donneurs d’électrons (bases), mais certains coordinats possèdent aussi, outre des orbitales 神 saturées, des orbitales 神 vacantes susceptibles d’accepter un transfert électronique du métal vers le coordinat. Dans cette donation en retour, le métal apparaît comme un donneur 神 et le coordinat comme un accepteur 神. La liaison est alors nettement renforcée par rapport à une interaction de type classique. C’est le cas des métaux carbonyles formulés de façon générale Mp (CO)r (cf. COORDINATION [chimie] - Chimie de coordination).On appelle coordinat unidenté un coordinat fixé en un «seul point» à l’ion métallique (eau: H2O; ammoniac: NH3, etc.). Les molécules ou les ions possédant deux ou plusieurs atomes donneurs sont susceptibles de former deux, trois, quatre... liaisons de coordination. De tels ligands sont alors bidentés, tridentés, quadridentés et plus généralement multidentés. Le nombre d’atomes donneurs liés à l’ion métallique définit le nombre de coordination de l’atome métallique central. Un métal peut présenter plusieurs nombres de coordination caractéristiques de la valence de l’atome central ou de la nature du coordinat envisagé. La notion de nombre de coordination est différente dans le cas des structures cristallines dans lesquelles il est déterminé par le nombre de voisins proches d’un atome dans le cristal et dépend par exemple du rapport des rayons ioniques (6 dans le chlorure de sodium, NaCl, et 8 dans le chlorure de césium, CsCl).La stéréochimie des complexes est déterminée en grande partie par le nombre de coordination qui varie de 2 à 9 (quelques cas de coordinence 10 et 11 ont été décrits) avec 4 et 6 comme valeurs les plus fréquentes. De très nombreux types de configurations géométriques sont alors à envisager et leur étude exhaustive dépasse le cadre de cette contribution. La coordinence 4 implique en général une structure tétraédrique, ou carrée plane, et la coordinence 6 une structure octaédrique, éventuellement distordue (effet Jahn-Teller).Dans une solution où coexistent le cation métallique solvaté et deux coordinats différents A et B, le complexe Mp Hq Ar Bs , supposé soluble, est formé suivant l’équilibre: p M + q H + r A + s B 曆 Mp Hq Ar Bs (les charges sont omises afin d’alléger l’écriture).Cette notation recouvre la plupart des cas de coordination que nous envisageons. La constante de stabilité ionique globale 廓pqrs de l’espèce Mp Hq Ar Bs est définie par:
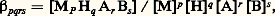 où les termes entre crochets désignent des concentrations en moles par litre. Au dénominateur figurent les concentrations libres (espèces non liées) des ions métalliques, des protons et des coordinats. La stabilité ionique du complexe est donnée par la valeur de log10 廓pqrs , indiquée dans les tables de constantes spécialisées. Le complexe sera d’autant plus stable que la valeur de log10 廓pqrs sera élevée. L’équilibre ci-dessus sera alors déplacé vers la droite, entraînant la formation du complexe.Ces constantes sont déterminées expérimentalement à partir de mesures électrochimiques (potentiels d’électrodes de pH et d’électrodes spécifiques, polarographie, chronopotentiométrie, etc.), de mesures optiques (spectroscopie dans l’ultraviolet et le visible et plus rarement dans l’infrarouge, Raman, résonance magnétique nucléaire, polarimétrie, dichroïsme circulaire) ou de mesures calorimétriques.Dans la plupart des cas, l’exploitation des données expérimentales nécessite l’utilisation de méthodes de traitements informatisées. Ces méthodes, devenues très accessibles depuis le développement de la micro-informatique, permettent l’analyse des équilibres de complexation en solution grâce à de nombreux programmes de calcul développés d’après les travaux originaux du groupe de L. G. Sillen (1916-1970) à Stockholm. Ces techniques permettent le tracé des diagrammes de répartition des espèces en solution (fig. 1), et cet excellent moyen de visualisation de l’évolution des phénomènes de complexation est largement employé lors de calculs de simulation concernant les équilibres multimétaux-multicoordinats.2. Différents cas de coordinationComplexes mononucléairesIl s’agit des complexes contenant un seul ion métallique central (p = 1 dans la notation Mp Hq Ar Bs ). Si ces complexes comportent un seul coordinat (r ou s = 0), on obtient des complexes simples binaires du type Co(NH3)63 +, AlCl-4, Fe(CN)63-, Cr(CO)6... Dans ces cas, les méthodes classiques de détermination des constantes de stabilité ionique (par exemple: méthode de Bjerrum) s’appliquent aisément.ChélatesDans tous les cas, le terme complexe est remplacé par le terme chélate (du grec Khêlê , pince) lorsqu’un coordinat multidenté peut se fixer au métal par deux ou plusieurs «dents» en formant une structure cyclique dont le schéma de la figure 2 explicite seulement quelques possibilités. De tels coordinats sont des agents chélatants. A. Werner (1901), puis H. Ley (1904) avaient déjà mentionné l’existence de certains «complexes internes» du platine et du cuivre et reconnu leur structure cyclique (fig. 2). C’est en 1920 que G. T. Morgan et D. H. K. Drew introduisent la terminologie plus précise et plus imagée que nous avons indiquée. Les chélates se distinguent essentiellement des autres catégories de complexes par leur grande stabilité (ionique) due à l’existence d’un (ou de plusieurs) cycle(s) dans leur structure.L’effet chélateL’exaltation de la stabilité d’une espèce complexe par formation d’un ou de plusieurs cycles de chélation porte le nom d’«effet chélate» (G. Schwarzenbach, 1952). Ainsi le cation complexe [Ni(NH3)2 (H2O)4]2+ (lg 廓 力 4,9) est nettement moins stable que le cation chélate [Ni en (H2O)4]2+ (lg 廓 力 7,3) dans lequel les deux coordinats monodentés NH3 ont été remplacés par un coordinat bidenté en 略 éthylènediamine: NH2 漣CH2 漣CH2 漣NH2. Il y a eu, ici, création d’un cycle à cinq maillons sur le cation Ni2+ (fig. 2). Cet effet chélate résulte essentiellement d’un effet entropique facile à expliquer qualitativement. La formation du cation complexe provoque la disparition de deux espèces indépendantes alors que celle du cation chélate ne provoque la disparition que d’une seule espèce indépendante, induisant un accroissement relatif d’entropie pour la réaction de chélation, donc un accroissement de stabilité. Il est alors facile d’en conclure que les chélates seront d’autant plus stables que le nombre de cycles qu’ils comportent sera plus élevé; on en trouve une confirmation dans l’extraordinaire stabilité des porphyrines (4 cycles) et dans l’aptitude particulière de l’acide éthylène-diamine-tétra-acétique (EDTA), coordinat hexadenté, à «séquestrer» fortement la plupart des ions métalliques (de 3 à 5 cycles selon la coordinence de l’ion), d’où son utilisation en complexométrie:
où les termes entre crochets désignent des concentrations en moles par litre. Au dénominateur figurent les concentrations libres (espèces non liées) des ions métalliques, des protons et des coordinats. La stabilité ionique du complexe est donnée par la valeur de log10 廓pqrs , indiquée dans les tables de constantes spécialisées. Le complexe sera d’autant plus stable que la valeur de log10 廓pqrs sera élevée. L’équilibre ci-dessus sera alors déplacé vers la droite, entraînant la formation du complexe.Ces constantes sont déterminées expérimentalement à partir de mesures électrochimiques (potentiels d’électrodes de pH et d’électrodes spécifiques, polarographie, chronopotentiométrie, etc.), de mesures optiques (spectroscopie dans l’ultraviolet et le visible et plus rarement dans l’infrarouge, Raman, résonance magnétique nucléaire, polarimétrie, dichroïsme circulaire) ou de mesures calorimétriques.Dans la plupart des cas, l’exploitation des données expérimentales nécessite l’utilisation de méthodes de traitements informatisées. Ces méthodes, devenues très accessibles depuis le développement de la micro-informatique, permettent l’analyse des équilibres de complexation en solution grâce à de nombreux programmes de calcul développés d’après les travaux originaux du groupe de L. G. Sillen (1916-1970) à Stockholm. Ces techniques permettent le tracé des diagrammes de répartition des espèces en solution (fig. 1), et cet excellent moyen de visualisation de l’évolution des phénomènes de complexation est largement employé lors de calculs de simulation concernant les équilibres multimétaux-multicoordinats.2. Différents cas de coordinationComplexes mononucléairesIl s’agit des complexes contenant un seul ion métallique central (p = 1 dans la notation Mp Hq Ar Bs ). Si ces complexes comportent un seul coordinat (r ou s = 0), on obtient des complexes simples binaires du type Co(NH3)63 +, AlCl-4, Fe(CN)63-, Cr(CO)6... Dans ces cas, les méthodes classiques de détermination des constantes de stabilité ionique (par exemple: méthode de Bjerrum) s’appliquent aisément.ChélatesDans tous les cas, le terme complexe est remplacé par le terme chélate (du grec Khêlê , pince) lorsqu’un coordinat multidenté peut se fixer au métal par deux ou plusieurs «dents» en formant une structure cyclique dont le schéma de la figure 2 explicite seulement quelques possibilités. De tels coordinats sont des agents chélatants. A. Werner (1901), puis H. Ley (1904) avaient déjà mentionné l’existence de certains «complexes internes» du platine et du cuivre et reconnu leur structure cyclique (fig. 2). C’est en 1920 que G. T. Morgan et D. H. K. Drew introduisent la terminologie plus précise et plus imagée que nous avons indiquée. Les chélates se distinguent essentiellement des autres catégories de complexes par leur grande stabilité (ionique) due à l’existence d’un (ou de plusieurs) cycle(s) dans leur structure.L’effet chélateL’exaltation de la stabilité d’une espèce complexe par formation d’un ou de plusieurs cycles de chélation porte le nom d’«effet chélate» (G. Schwarzenbach, 1952). Ainsi le cation complexe [Ni(NH3)2 (H2O)4]2+ (lg 廓 力 4,9) est nettement moins stable que le cation chélate [Ni en (H2O)4]2+ (lg 廓 力 7,3) dans lequel les deux coordinats monodentés NH3 ont été remplacés par un coordinat bidenté en 略 éthylènediamine: NH2 漣CH2 漣CH2 漣NH2. Il y a eu, ici, création d’un cycle à cinq maillons sur le cation Ni2+ (fig. 2). Cet effet chélate résulte essentiellement d’un effet entropique facile à expliquer qualitativement. La formation du cation complexe provoque la disparition de deux espèces indépendantes alors que celle du cation chélate ne provoque la disparition que d’une seule espèce indépendante, induisant un accroissement relatif d’entropie pour la réaction de chélation, donc un accroissement de stabilité. Il est alors facile d’en conclure que les chélates seront d’autant plus stables que le nombre de cycles qu’ils comportent sera plus élevé; on en trouve une confirmation dans l’extraordinaire stabilité des porphyrines (4 cycles) et dans l’aptitude particulière de l’acide éthylène-diamine-tétra-acétique (EDTA), coordinat hexadenté, à «séquestrer» fortement la plupart des ions métalliques (de 3 à 5 cycles selon la coordinence de l’ion), d’où son utilisation en complexométrie: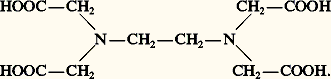 L’EDTA est devenu rapidement un produit industriel important. Il est le chef de file de toute une série d’acides aminopolycarboxyliques qui ont des propriétés voisines quant à la chélation des cations métalliques.Le nombre de maillons d’un cycle de chélation dépend évidemment de la nature du coordinat; comme on pouvait s’y attendre, ce sont, comme pour les composés organiques, les cycles à cinq ou six maillons qui présentent le maximum de stabilité. Précisons cependant que les chélates les plus stables comportent des cycles à cinq maillons pour les coordinats saturés et à six maillons pour les coordinats non saturés.Complexes polynucléairesCes composés peuvent contenir deux ou plusieurs ions centraux (p 礪 1 dans Mp Hq Ar Bs ) comme Co2 (CO)8, Mn2(CO)10. Il peut y avoir une ou des liaison(s) métalmétal ou un pontage des ions métalliques par les coordinats comme dans l’ion hexaamino- 猪-amido- 猪-dihydroxodicobalt (III) [fig. 3]. Un cas très intéressant concerne les clusters , qui sont des molécules où chaque atome métallique est directement lié à au moins deux autres atomes métalliques. Le cas le plus simple des clusters moléculaires est le triosmiumdodécacarbonyle Os3(CO)12. On connaît aussi des clusters hétéronucléaires du type FeRu2Os(CO)13H2 et FeRuOs2(CO)13H2, où le «cœur» métallique associe plusieurs (ici 3) métaux différents. Ces clusters constituent actuellement une nouvelle voie d’études fondamentales et appliquées (liaisons métal-métal, coordinations non usuelles des ligands, mécanisme de catalyse homogène et hétérogène; (cf. chimie de COORDINATION).Les polyanions minéraux sont aussi des espèces polynucléaires; de tels composés V274-, V393-, V4124-, V10286- sont obtenus, par exemple, en acidifiant les solutions alcalines d’orthovanadate V43- .Complexes protonés et hydroxydésUne valeur de q positive ou négative dans la formule générale Mp Hq Ar Bs indique respectivement l’existence de complexes protonés ou hydroxydés. Dans ce dernier cas, il s’agit de phénomènes d’hydrolyse intéressant notamment les métaux de degré d’oxydation élevé, ayant un fort caractère acide, tels Fe(III), Al(III), Ti(IV), Th(IV)... L’espèce Fe2H-24+ [notation identique à Fe2(OH)24+] est un exemple de ces complexes hydroxydés qui sont souvent aussi polynucléaires. Il est important de savoir que la proportion de complexes polynucléaires en solution décroît avec la dilution. Aussi, lorsque l’on considère l’hydrolyse d’un ion métallique, on peut négliger les équilibres de complexes polynucléaires, souvent mal connus, si la concentration de l’ion métallique est suffisamment faible, ce que l’on obtient par dilution ou addition d’un agent complexant entrant en compétition avec les ions hydroxyde.Complexes mixtesLorsqu’on fixe sur un même ion métallique plusieurs coordinats de nature différente, on obtient un complexe mixte (appelé complexe mixte ternaire s’il possède seulement deux types de coordinats différents A et B). Les études effectuées sur ces espèces ont démontré la surstabilisation des complexes mixtes par rapport aux complexes simples binaires, que l’on peut apprécier par exemple au moyen de la constante d’équilibre de la réaction:
L’EDTA est devenu rapidement un produit industriel important. Il est le chef de file de toute une série d’acides aminopolycarboxyliques qui ont des propriétés voisines quant à la chélation des cations métalliques.Le nombre de maillons d’un cycle de chélation dépend évidemment de la nature du coordinat; comme on pouvait s’y attendre, ce sont, comme pour les composés organiques, les cycles à cinq ou six maillons qui présentent le maximum de stabilité. Précisons cependant que les chélates les plus stables comportent des cycles à cinq maillons pour les coordinats saturés et à six maillons pour les coordinats non saturés.Complexes polynucléairesCes composés peuvent contenir deux ou plusieurs ions centraux (p 礪 1 dans Mp Hq Ar Bs ) comme Co2 (CO)8, Mn2(CO)10. Il peut y avoir une ou des liaison(s) métalmétal ou un pontage des ions métalliques par les coordinats comme dans l’ion hexaamino- 猪-amido- 猪-dihydroxodicobalt (III) [fig. 3]. Un cas très intéressant concerne les clusters , qui sont des molécules où chaque atome métallique est directement lié à au moins deux autres atomes métalliques. Le cas le plus simple des clusters moléculaires est le triosmiumdodécacarbonyle Os3(CO)12. On connaît aussi des clusters hétéronucléaires du type FeRu2Os(CO)13H2 et FeRuOs2(CO)13H2, où le «cœur» métallique associe plusieurs (ici 3) métaux différents. Ces clusters constituent actuellement une nouvelle voie d’études fondamentales et appliquées (liaisons métal-métal, coordinations non usuelles des ligands, mécanisme de catalyse homogène et hétérogène; (cf. chimie de COORDINATION).Les polyanions minéraux sont aussi des espèces polynucléaires; de tels composés V274-, V393-, V4124-, V10286- sont obtenus, par exemple, en acidifiant les solutions alcalines d’orthovanadate V43- .Complexes protonés et hydroxydésUne valeur de q positive ou négative dans la formule générale Mp Hq Ar Bs indique respectivement l’existence de complexes protonés ou hydroxydés. Dans ce dernier cas, il s’agit de phénomènes d’hydrolyse intéressant notamment les métaux de degré d’oxydation élevé, ayant un fort caractère acide, tels Fe(III), Al(III), Ti(IV), Th(IV)... L’espèce Fe2H-24+ [notation identique à Fe2(OH)24+] est un exemple de ces complexes hydroxydés qui sont souvent aussi polynucléaires. Il est important de savoir que la proportion de complexes polynucléaires en solution décroît avec la dilution. Aussi, lorsque l’on considère l’hydrolyse d’un ion métallique, on peut négliger les équilibres de complexes polynucléaires, souvent mal connus, si la concentration de l’ion métallique est suffisamment faible, ce que l’on obtient par dilution ou addition d’un agent complexant entrant en compétition avec les ions hydroxyde.Complexes mixtesLorsqu’on fixe sur un même ion métallique plusieurs coordinats de nature différente, on obtient un complexe mixte (appelé complexe mixte ternaire s’il possède seulement deux types de coordinats différents A et B). Les études effectuées sur ces espèces ont démontré la surstabilisation des complexes mixtes par rapport aux complexes simples binaires, que l’on peut apprécier par exemple au moyen de la constante d’équilibre de la réaction: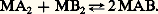 Cet équilibre est déplacé vers la droite en faveur de la formation de l’espèce mixte MAB. L’origine de cette surstabilisation est essentiellement entropique. Elle s’explique aisément par des considérations statistiques mais néanmoins d’autres facteurs interviennent. Il ne fait aucun doute que cette formation préférentielle de complexes mixtes ne doit pas être négligée dans l’analyse complexométrique, dans les techniques d’extraction par solvant (interprétation des phénomènes de synergisme), mais surtout dans l’étude des équilibres en chimie bio-inorganique où les ions métalliques présents dans les organismes vivants se trouvent, en règle générale, en présence de plusieurs coordinats potentiels différents.Coordinats macrocycliquesDe nouveaux coordinats, dont les applications potentielles sont nombreuses, ont été synthétisés récemment et conduisent à des complexes doués de propriétés remarquables (par exemple, solubiliser le permanganate de potassium dans des solvants non polaires comme le benzène!). En effet, il est connu que certaines substances naturelles macrocycliques, tels certains antibiotiques comme la valinomycine, complexent (sélectivement pour certains) les cations alcalins souvent considérés comme résistant habituellement à la complexation. De plus, ces substances permettent le transport sélectif de cations complexés à travers des membranes artificielles ou biologiques. Plusieurs équipes de recherche ont alors tenté d’obtenir des macrocycles synthétiques susceptibles de présenter des propriétés analogues. Des études systématiques ont été menées principalement sur deux familles de composés (fig. 4):– les polyéthers macrocycliques «crowns ou éthers-couronnes» (C. J. Pedersen, 1967);– les diaza-polyoxa-macrobicycles appelés communément cryptates (du latin Crypta , du grec Kruptos , caché; J. M. Lehn, 1969) pour indiquer leur nature structurale particulière; le cation se situant à l’intérieur d’une cavité macrocyclique. Il est possible de synthétiser des coordinats «sur mesure» ayant des propriétés de coordination sélective en fixant la dimension, la nature chimique et géométrique du macrocycle. De même, l’extérieur du macrocycle, essentiellement hydrophobe, peut être modifié de façon à faciliter la dissolution de sels dans les solvants organiques provoquant ainsi une augmentation de réactivité des anions.Effet macrocycliqueL’effet macrocyclique rend compte de la stabilité ionique accrue d’un complexe métallique formé avec un coordinat multidenté cyclique par rapport à celle du complexe du même cation métallique avec un coordinat comparable mais acyclique; c’est ainsi que le complexe de Zn2+ avec le 1,4,8,11-tétra-azacyclo-tétra-décane
Cet équilibre est déplacé vers la droite en faveur de la formation de l’espèce mixte MAB. L’origine de cette surstabilisation est essentiellement entropique. Elle s’explique aisément par des considérations statistiques mais néanmoins d’autres facteurs interviennent. Il ne fait aucun doute que cette formation préférentielle de complexes mixtes ne doit pas être négligée dans l’analyse complexométrique, dans les techniques d’extraction par solvant (interprétation des phénomènes de synergisme), mais surtout dans l’étude des équilibres en chimie bio-inorganique où les ions métalliques présents dans les organismes vivants se trouvent, en règle générale, en présence de plusieurs coordinats potentiels différents.Coordinats macrocycliquesDe nouveaux coordinats, dont les applications potentielles sont nombreuses, ont été synthétisés récemment et conduisent à des complexes doués de propriétés remarquables (par exemple, solubiliser le permanganate de potassium dans des solvants non polaires comme le benzène!). En effet, il est connu que certaines substances naturelles macrocycliques, tels certains antibiotiques comme la valinomycine, complexent (sélectivement pour certains) les cations alcalins souvent considérés comme résistant habituellement à la complexation. De plus, ces substances permettent le transport sélectif de cations complexés à travers des membranes artificielles ou biologiques. Plusieurs équipes de recherche ont alors tenté d’obtenir des macrocycles synthétiques susceptibles de présenter des propriétés analogues. Des études systématiques ont été menées principalement sur deux familles de composés (fig. 4):– les polyéthers macrocycliques «crowns ou éthers-couronnes» (C. J. Pedersen, 1967);– les diaza-polyoxa-macrobicycles appelés communément cryptates (du latin Crypta , du grec Kruptos , caché; J. M. Lehn, 1969) pour indiquer leur nature structurale particulière; le cation se situant à l’intérieur d’une cavité macrocyclique. Il est possible de synthétiser des coordinats «sur mesure» ayant des propriétés de coordination sélective en fixant la dimension, la nature chimique et géométrique du macrocycle. De même, l’extérieur du macrocycle, essentiellement hydrophobe, peut être modifié de façon à faciliter la dissolution de sels dans les solvants organiques provoquant ainsi une augmentation de réactivité des anions.Effet macrocycliqueL’effet macrocyclique rend compte de la stabilité ionique accrue d’un complexe métallique formé avec un coordinat multidenté cyclique par rapport à celle du complexe du même cation métallique avec un coordinat comparable mais acyclique; c’est ainsi que le complexe de Zn2+ avec le 1,4,8,11-tétra-azacyclo-tétra-décane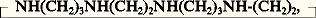 C10H244, possède une constante de stabilité lg 廓 = 15,34 alors que, pour le complexe de Zn2+ avec le coordinat acyclique 1,2-bis (3-aminopropylamino)éthane (NH2(CH2)3NH(CH2)-)2, C8H224, on détermine lg 廓 = 11,25. Contrairement à l’effet chélate qui s’avère comme étant essentiellement d’origine entropique, l’effet macrocyclique est à la fois d’origine enthalpique et entropique.PolychélatesLes créations de ponts entre les molécules coordinantes, à l’aide d’ions métalliques, permettent d’aboutir à des enchaînements moléculaires importants. Ces derniers sont alors désignés sous le nom de «polymères» de coordination, dont les représentants les plus intéressants sont constitués par les polychélates. C’est ainsi que, en faisant réagir un coordinat comportant deux groupes bidentés sur un ion de coordinence 4, on pourra obtenir un polymère linéaire (fig. 5). Les polymères seront au contraire réticulés lorsque de tels coordinats se fixeront sur des ions de coordinence 6.Ces polychélates ne présentent généralement pas les propriétés plastiques des polymères purement organiques, et leur insolubilité dans les solvants minéraux ou organiques n’en facilite pas l’étude. Certains d’entre eux manifestent une stabilité thermique intéressante, et des recherches approfondies sur leurs autres propriétés physico-chimiques (conductivité électrique, propriétés magnétiques, optiques, etc.) peuvent conduire à des applications prometteuses.Coordinats polymèresAu lieu de former un polymère métallisé suivant le processus précédent, on peut envisager la complexation directe des ions métalliques par des polyélectrolytes. C’est ainsi que les substances humiques, qui constituent la composante organique principale des sols et des eaux, sont capables de former des complexes métalliques. Bien que la structure de ces acides polymères (acides humiques et fulviques) soit compliquée, quelques travaux ont permis de connaître la stabilité ionique de leurs complexes et de montrer, dans certains cas, une coordination de type «salicylate». Tous les polymères chélatants naturels se rangent dans cette rubrique: polyuronates (notamment pectine et acide alginique), chitine, sulfate de chondroïtine, acide hyaluronique, etc.3. Principaux domaines d’applicationL’intérêt suscité par la chimie des complexes provient de la diversité de leurs très nombreuses applications qui ne cessent de se développer dans tous les domaines de la chimie, débordant même sur la physique, la métallurgie, la chimie bio-inorganique.L’importance industrielle réside tout d’abord dans la catalyse homogène où l’utilisation de catalyseurs organométalliques (composés à liaison(s) métal-carbone) ou de sels ou de complexes solubles de métaux de transition offre les avantages d’une plus grande efficacité, d’une bonne sélectivité et de conditions de réactions plus douces qu’en catalyse hétérogène. De plus, les mécanismes réactionnels peuvent être analysés (et donc améliorés) au moyen des techniques spectroscopiques et cinétiques modernes. Plus de vingt procédés industriels utilisent des complexes métalliques solubles comme catalyseurs dans des réactions d’hydrogénation et d’oxydation des hydrocarbures, de polymérisation des oléfines, de synthèses d’acides, d’aldéhydes et d’alcools (cf. CATALYSE Catalyse homogène).De nombreux complexes utilisés comme précurseurs conduisent, après pyrolyse dans des atmosphères gazeuses appropriées, à l’obtention de phases solides (oxydes, sulfures, nitrures, carbures, etc.) finement divisées.La chimie analytique met à profit l’insolubilité de certains complexes pour des dosages gravimétriques, leur coloration spécifique pour la détection ou le dosage absorptiométrique des ions métalliques, mais, surtout, elle dispose d’un très grand nombre de méthodes complexométriques pour doser et séparer les ions. Pour une utilisation rationnelle des complexes en chimie analytique, il importe alors de connaître non seulement les constantes de stabilité ionique définies ci-dessus, mais aussi les constantes «conditionnelles» tenant compte de l’influence de l’acidité de la solution, mais aussi des ions métalliques ou des coordinats parasites... (A. Ringbom, 1967).Les agents complexants sont largement utilisés dans les opérations relatives à la chimie minérale. La détermination de la dureté de l’eau aussi bien que son adoucissement s’effectuent classiquement au moyen de séquestrants organiques ou minéraux, comme dans le cas du tripolyphosphate de sodium Na5P310. Des coordinats polymères sont utilisés pour éliminer les cations métalliques des eaux résiduaires par coagulation-floculation. En outre, les séparations très délicates des éléments du groupe des lanthanides et du groupe des actinides n’ont pu être réalisées commodément que selon deux techniques utilisées soit séparément, soit conjointement:– le fractionnement sur échangeurs d’ions faisant appel aux séquestrants organiques comme agent d’élution;– l’extraction méthodique liquide-liquide fondée sur la formation de complexes solubles en milieu organique; c’est ainsi que sont séparés industriellement les métaux des terres rares au moyen d’agents extractants à propriétés complexantes, chélatantes ou solvatantes tels que le tri(n-butyl)phosphate (TBP); le retraitement des combustibles nucléaires utilise aussi ces techniques.La métallurgie emploie des complexes dans certaines opérations d’enrichissement de minerais (flottation) ou de séparation [procédé Mond de purification du nickel par formation de Ni(CO)4].Les plus anciens teinturiers, faisant appel aux laques d’alizarine telles que le fameux rouge turc, ignoraient certes la nature de ces composés chélates mais en connaissaient bien la solidité. La chimie tinctoriale, devant satisfaire les exigences sans cesse croissantes imposées à ses colorants (variété, éclat, stabilité), développa les colorants à complexe métallique (ou métallifères) utilisés dans les opérations de métallisation. De même, un pigment classique comme la phtalocyanine est un chélate du cuivre: c’est le pigment bleu par excellence.Dans le domaine phytosanitaire, ce sont surtout des carences en métaux qui sont éliminées par l’utilisation de complexes: c’est ainsi que les traitements antichlorose mettent en œuvre des chélates de fer(III) de très grande stabilité ionique.On ne saurait mieux faire apprécier l’importance des chélates dans le règne végétal qu’en rappelant leur rôle essentiel dans les processus fondamentaux tels que la photosynthèse initiée par la chlorophylle (en réalité, il existe plusieurs types de chlorophylles qui ont toutes en commun un chromophore constitué par la jonction de quatre noyaux pyrroles en un système cyclique plan chélatant l’ion central Mg2+). Des porphyrines analogues peuvent aussi être utilisées dans des processus de stockage d’énergie (cytochromes), comme catalyseurs enzymatiques (catalase) ou encore comme transporteur d’oxygène (hémoglobine).Le domaine de la chimie bio-inorganique, après avoir été longtemps négligé, connaît actuellement un développement rapide étant donné l’importance des ions métalliques dans les fonctions vitales des organismes vivants. C’est ainsi que sont engagées de multiples recherches concernant la réactivité, la synthèse, la stabilité, la structure et la formation des complexes et chélates d’intérêt biologique. Il est certain que tous les éléments métalliques rencontrés dans la matière vivante animale ou végétale font généralement partie de cycles de chélation. Une concentration anormale de certains d’entre eux entraîne le plus souvent des perturbations dans les mécanismes cellulaires. En contrepartie, la thérapeutique chimique utilise fréquemment la complexation ou la chélation comme moyen d’introduction ou d’élimination des éléments métalliques dans l’organisme: utilisation du cis -dichlorodiamine platine (II), Pt C12 (NH3)2 lors de traitements anticancéreux; élimination du cuivre (II) dans la maladie de Wilson par la D-pénicillamine, etc. De telles applications impliquent une connaissance très approfondie des coordinats nombreux, variés et de structure compliquée, que nous offre la matière vivante, ainsi qu’une parfaite maîtrise des équilibres physico-chimiques où interviennent ces complexes.
C10H244, possède une constante de stabilité lg 廓 = 15,34 alors que, pour le complexe de Zn2+ avec le coordinat acyclique 1,2-bis (3-aminopropylamino)éthane (NH2(CH2)3NH(CH2)-)2, C8H224, on détermine lg 廓 = 11,25. Contrairement à l’effet chélate qui s’avère comme étant essentiellement d’origine entropique, l’effet macrocyclique est à la fois d’origine enthalpique et entropique.PolychélatesLes créations de ponts entre les molécules coordinantes, à l’aide d’ions métalliques, permettent d’aboutir à des enchaînements moléculaires importants. Ces derniers sont alors désignés sous le nom de «polymères» de coordination, dont les représentants les plus intéressants sont constitués par les polychélates. C’est ainsi que, en faisant réagir un coordinat comportant deux groupes bidentés sur un ion de coordinence 4, on pourra obtenir un polymère linéaire (fig. 5). Les polymères seront au contraire réticulés lorsque de tels coordinats se fixeront sur des ions de coordinence 6.Ces polychélates ne présentent généralement pas les propriétés plastiques des polymères purement organiques, et leur insolubilité dans les solvants minéraux ou organiques n’en facilite pas l’étude. Certains d’entre eux manifestent une stabilité thermique intéressante, et des recherches approfondies sur leurs autres propriétés physico-chimiques (conductivité électrique, propriétés magnétiques, optiques, etc.) peuvent conduire à des applications prometteuses.Coordinats polymèresAu lieu de former un polymère métallisé suivant le processus précédent, on peut envisager la complexation directe des ions métalliques par des polyélectrolytes. C’est ainsi que les substances humiques, qui constituent la composante organique principale des sols et des eaux, sont capables de former des complexes métalliques. Bien que la structure de ces acides polymères (acides humiques et fulviques) soit compliquée, quelques travaux ont permis de connaître la stabilité ionique de leurs complexes et de montrer, dans certains cas, une coordination de type «salicylate». Tous les polymères chélatants naturels se rangent dans cette rubrique: polyuronates (notamment pectine et acide alginique), chitine, sulfate de chondroïtine, acide hyaluronique, etc.3. Principaux domaines d’applicationL’intérêt suscité par la chimie des complexes provient de la diversité de leurs très nombreuses applications qui ne cessent de se développer dans tous les domaines de la chimie, débordant même sur la physique, la métallurgie, la chimie bio-inorganique.L’importance industrielle réside tout d’abord dans la catalyse homogène où l’utilisation de catalyseurs organométalliques (composés à liaison(s) métal-carbone) ou de sels ou de complexes solubles de métaux de transition offre les avantages d’une plus grande efficacité, d’une bonne sélectivité et de conditions de réactions plus douces qu’en catalyse hétérogène. De plus, les mécanismes réactionnels peuvent être analysés (et donc améliorés) au moyen des techniques spectroscopiques et cinétiques modernes. Plus de vingt procédés industriels utilisent des complexes métalliques solubles comme catalyseurs dans des réactions d’hydrogénation et d’oxydation des hydrocarbures, de polymérisation des oléfines, de synthèses d’acides, d’aldéhydes et d’alcools (cf. CATALYSE Catalyse homogène).De nombreux complexes utilisés comme précurseurs conduisent, après pyrolyse dans des atmosphères gazeuses appropriées, à l’obtention de phases solides (oxydes, sulfures, nitrures, carbures, etc.) finement divisées.La chimie analytique met à profit l’insolubilité de certains complexes pour des dosages gravimétriques, leur coloration spécifique pour la détection ou le dosage absorptiométrique des ions métalliques, mais, surtout, elle dispose d’un très grand nombre de méthodes complexométriques pour doser et séparer les ions. Pour une utilisation rationnelle des complexes en chimie analytique, il importe alors de connaître non seulement les constantes de stabilité ionique définies ci-dessus, mais aussi les constantes «conditionnelles» tenant compte de l’influence de l’acidité de la solution, mais aussi des ions métalliques ou des coordinats parasites... (A. Ringbom, 1967).Les agents complexants sont largement utilisés dans les opérations relatives à la chimie minérale. La détermination de la dureté de l’eau aussi bien que son adoucissement s’effectuent classiquement au moyen de séquestrants organiques ou minéraux, comme dans le cas du tripolyphosphate de sodium Na5P310. Des coordinats polymères sont utilisés pour éliminer les cations métalliques des eaux résiduaires par coagulation-floculation. En outre, les séparations très délicates des éléments du groupe des lanthanides et du groupe des actinides n’ont pu être réalisées commodément que selon deux techniques utilisées soit séparément, soit conjointement:– le fractionnement sur échangeurs d’ions faisant appel aux séquestrants organiques comme agent d’élution;– l’extraction méthodique liquide-liquide fondée sur la formation de complexes solubles en milieu organique; c’est ainsi que sont séparés industriellement les métaux des terres rares au moyen d’agents extractants à propriétés complexantes, chélatantes ou solvatantes tels que le tri(n-butyl)phosphate (TBP); le retraitement des combustibles nucléaires utilise aussi ces techniques.La métallurgie emploie des complexes dans certaines opérations d’enrichissement de minerais (flottation) ou de séparation [procédé Mond de purification du nickel par formation de Ni(CO)4].Les plus anciens teinturiers, faisant appel aux laques d’alizarine telles que le fameux rouge turc, ignoraient certes la nature de ces composés chélates mais en connaissaient bien la solidité. La chimie tinctoriale, devant satisfaire les exigences sans cesse croissantes imposées à ses colorants (variété, éclat, stabilité), développa les colorants à complexe métallique (ou métallifères) utilisés dans les opérations de métallisation. De même, un pigment classique comme la phtalocyanine est un chélate du cuivre: c’est le pigment bleu par excellence.Dans le domaine phytosanitaire, ce sont surtout des carences en métaux qui sont éliminées par l’utilisation de complexes: c’est ainsi que les traitements antichlorose mettent en œuvre des chélates de fer(III) de très grande stabilité ionique.On ne saurait mieux faire apprécier l’importance des chélates dans le règne végétal qu’en rappelant leur rôle essentiel dans les processus fondamentaux tels que la photosynthèse initiée par la chlorophylle (en réalité, il existe plusieurs types de chlorophylles qui ont toutes en commun un chromophore constitué par la jonction de quatre noyaux pyrroles en un système cyclique plan chélatant l’ion central Mg2+). Des porphyrines analogues peuvent aussi être utilisées dans des processus de stockage d’énergie (cytochromes), comme catalyseurs enzymatiques (catalase) ou encore comme transporteur d’oxygène (hémoglobine).Le domaine de la chimie bio-inorganique, après avoir été longtemps négligé, connaît actuellement un développement rapide étant donné l’importance des ions métalliques dans les fonctions vitales des organismes vivants. C’est ainsi que sont engagées de multiples recherches concernant la réactivité, la synthèse, la stabilité, la structure et la formation des complexes et chélates d’intérêt biologique. Il est certain que tous les éléments métalliques rencontrés dans la matière vivante animale ou végétale font généralement partie de cycles de chélation. Une concentration anormale de certains d’entre eux entraîne le plus souvent des perturbations dans les mécanismes cellulaires. En contrepartie, la thérapeutique chimique utilise fréquemment la complexation ou la chélation comme moyen d’introduction ou d’élimination des éléments métalliques dans l’organisme: utilisation du cis -dichlorodiamine platine (II), Pt C12 (NH3)2 lors de traitements anticancéreux; élimination du cuivre (II) dans la maladie de Wilson par la D-pénicillamine, etc. De telles applications impliquent une connaissance très approfondie des coordinats nombreux, variés et de structure compliquée, que nous offre la matière vivante, ainsi qu’une parfaite maîtrise des équilibres physico-chimiques où interviennent ces complexes.
Encyclopédie Universelle. 2012.